
- Academic Editor
†These authors contributed equally.
Transforming growth factor-
The mitogen-activated protein kinase (MAPK) cascade is a cornerstone in
eukaryotic signal transduction, characterized by a hierarchy of three kinase
classes: MAP3K, MAP2K, and MAPK [1]. Among these, transforming growth
factor-
TAK1-binding proteins (TAB proteins), comprising TAB1, TAB2, and TAB3, are essential and specific
binding partners for TAK1. TAB1 binds to the N-terminus of TAK1, while TAB2 and
TAB3 associate with the C-terminus of TAK1 under stress-induced signaling [5].
The formation of the TAK1–TAB1–TAB2–TAB3 complex is pivotal for inducing the
autophosphorylation and subsequent activation of TAK1 [5]. This activation is
essential for the downstream activation of NF-
Ubiquitination, an omnipresent and versatile post-translational modification (PTM), plays a pivotal role in regulating the TAK–TAB complex, affecting its activation, stability, and degradation [8, 9]. This review dissects the mechanisms through which ubiquitination governs the TAK–TAB complex, shedding light on its contribution to cellular signaling and potential therapeutic avenues. A deeper understanding of these regulatory mechanisms enhances our knowledge of the TAK–TAB complex and creates new perspectives for targeting these pathways in disease contexts.
The TAK–TAB complex, central to cellular signaling, comprises the kinase TAK1 and its specific binding partners, TAB1, TAB2, and TAB3 [5]. Initial in vivo experiments revealed that global deletion of TAK1 results in early embryonic death due to defects in neural tube formation [10, 11]. Similarly, the elimination of TAK1’s essential activation partners, TAB1 and TAB2, leads to non-viability because of cardiovascular and liver malformations, respectively [12, 13, 14]. Researchers have employed tissue-specific conditional knockouts of TAK1 to overcome these challenges, gaining deeper insights into its role across different cell types. Such studies have shown that specific knockouts in immune cells disrupt B cell development and the immune response to antigens [11], while knockouts in non-immune cells such as the skin, liver, and intestinal cells cause aberrant development and inflammation [15], illustrating the complex and critical nature of TAK1 in both immune and non-immune cellular processes [11, 16].
TAK1 is a serine/threonine protein kinase featuring an N-terminal domain, a hinge region, and a C-terminal domain [17]. The N-terminal domain of TAK1 contains a conserved kinase domain, which is responsible for its enzymatic activity, phosphorylating specific serine and/or threonine residues on substrate proteins. ATP binds and transfers a phosphate group to the substrate in this kinase domain. The serine-rich and glycine-rich regions within this domain may regulate its kinase activity and play roles in protein–protein interactions (Fig. 1) [12, 16].
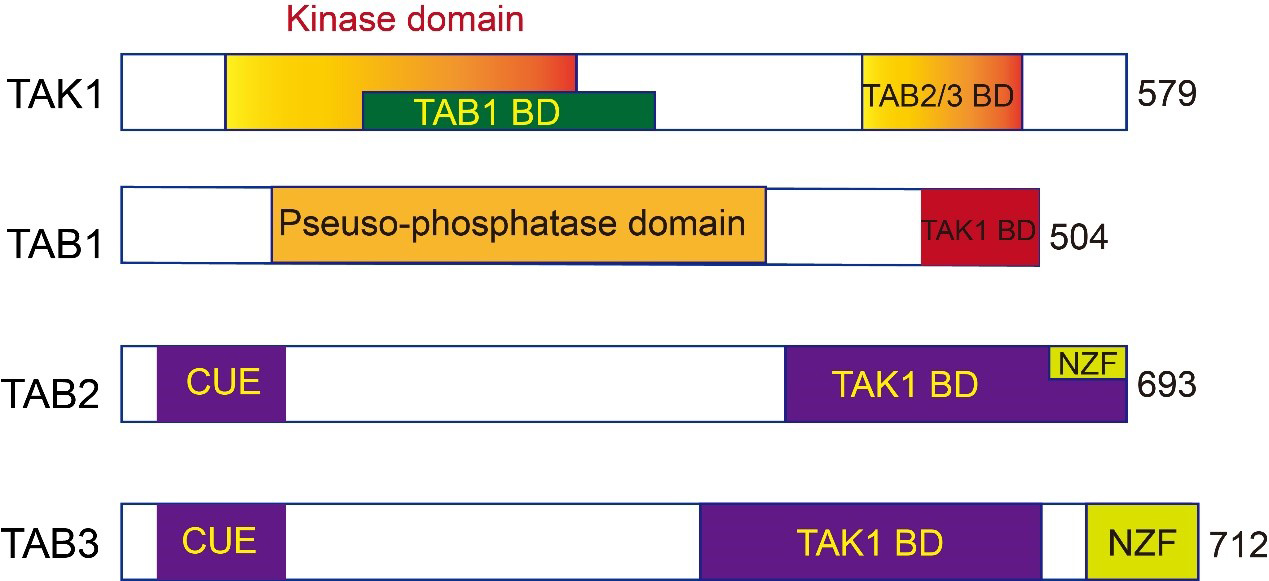 Fig. 1.
Fig. 1.Schematic illustration of the domain structures of human
transforming growth factor-
Research on TAK1 in mice has cast uncertainty on its viability as a therapeutic target. While no TAK1 loss-of-function mutations have been identified in humans, a gain-of-function mutation resulting from a truncated kinase form leads to developmental phenotypes, including craniofacial abnormalities [18]. This highlights the complex role of TAK1 in embryogenesis and development [19]. The discovery underscores the challenges in targeting kinases therapeutically, given the potential for genetic contradictions and the complexity of kinase-mediated signaling pathways, which may involve redundant or compensatory mechanisms [20]. Moreover, the scaffolding roles played by TAK1 further complicate the interpretation of its genetic studies, emphasizing that a nuanced approach is needed in pharmacologically manipulating TAK1 for therapeutic benefits.
The biological actions of TAK1 span various signaling pathways, and its activity
is regulated by diverse stimuli, including growth factors, cell adhesion, and
inflammatory factors [21]. Upon activation, TAK1 regulates downstream proteins,
including adaptor proteins in the NF-
Genome-wide association studies (GWAS) have placed TAK1 (MAP3K7) in the spotlight, underscoring its critical role in health and disease, thus marking it as a potential therapeutic target [23, 24, 25, 26, 27, 28]. Such studies have connected TAK1 with various diseases, including Crohn’s disease, ulcerative colitis, and inflammatory bowel disorder. In particular, GWAS on Japanese rheumatoid arthritis (RA) patients highlighted a significant association of TAK1 with susceptibility to diseases and response to anti-TNF treatments, suggesting its pivotal role in inflammatory disease management [25, 29]. The association of TAK1 with diverse conditions, from immune responses to genetic traits affecting biological functions, demonstrates its broad biological impact, emphasizing the need for further research into its mechanisms and therapeutic applications.
Given these diverse roles, TAK1 is considered a key molecular node in signaling networks that determine cell fate and function. Understanding the regulation of TAK1 and its pathways offers potential therapeutic targets for various diseases where its signaling is aberrant.
TAK1 binding proteins (TABs) serve as specific binding partners for TAK1 and include TAB1, TAB2, and TAB3. The activation of TAK1 relies on the assembly of these proteins, forming a complex that crucially regulates the kinase activity of TAK1 [5].
TAB1: Characterized by a pseudo-phosphatase domain and a TAK1 binding domain (TAK1 BD) (Fig. 1). The pseudo-phosphatase domain in TAB1 has a similar structure to protein phosphatase 2C (PP2C), despite lacking phosphatase activity. It interacts directly with the N-terminal kinase domain of TAK1 [30]. Disrupting this interaction can result in late-stage pregnancy edema and hemorrhage. In vitro experiments have found that TAB1 is indispensable for TAK1 activation through autophosphorylation [5, 31].
TAB2 and TAB3: Homologous proteins that share 48% of their amino acid sequences, indicating a significant evolutionary relationship. They feature an N-terminal coupling of ubiquitin to ER degradation (CUE) domain, a C-terminal TAK1 binding domain, and a Npl4 zinc-finger (NZF) domain [14, 16] (Fig. 1). The TAK1 binding domain in TAB2 and TAB3 is primarily responsible for binding with TAK1, while the CUE domain binds directly to ubiquitin, linking ubiquitinated proteins to endoplasmic reticulum degradation [32]. The NZF domain is responsible for binding polyubiquitin chains, specifically recognizing the polyubiquitin chains linked by lysine (K)63, crucial for TAK1 activation [33]. Although structurally similar, TAB2 and TAB3 can complement each other in certain functions. TAB2 gene knockout results in embryonic death in mice, while TAB3 gene knockout mice can be born normally, suggesting specific roles in regulating TAK1 activity [14, 16].
Through their binding domains, TAK1 and TAB1 are associated in unstimulated cells [31]. Upon stimulation, TAB2 and TAB3 bind to the C-terminal region of TAK1 through their respective binding domains, forming the TAK1–TAB1–TAB2/TAB3 complex. The functional assembly of TAK1 with TAB proteins in response to cellular stimuli underscores the complex’s versatility in regulating various biological processes. This interaction activates critical signaling pathways and reflects the complex’s adaptability to diverse cellular contexts, from immune responses to stress responses [12]. The detailed study of the TAK–TAB complex reveals its fundamental role in cellular signaling and disease mechanisms, presenting opportunities for therapeutic intervention.
TAK1, a pivotal multifunctional kinase, is a
critical upstream regulator in the NF-
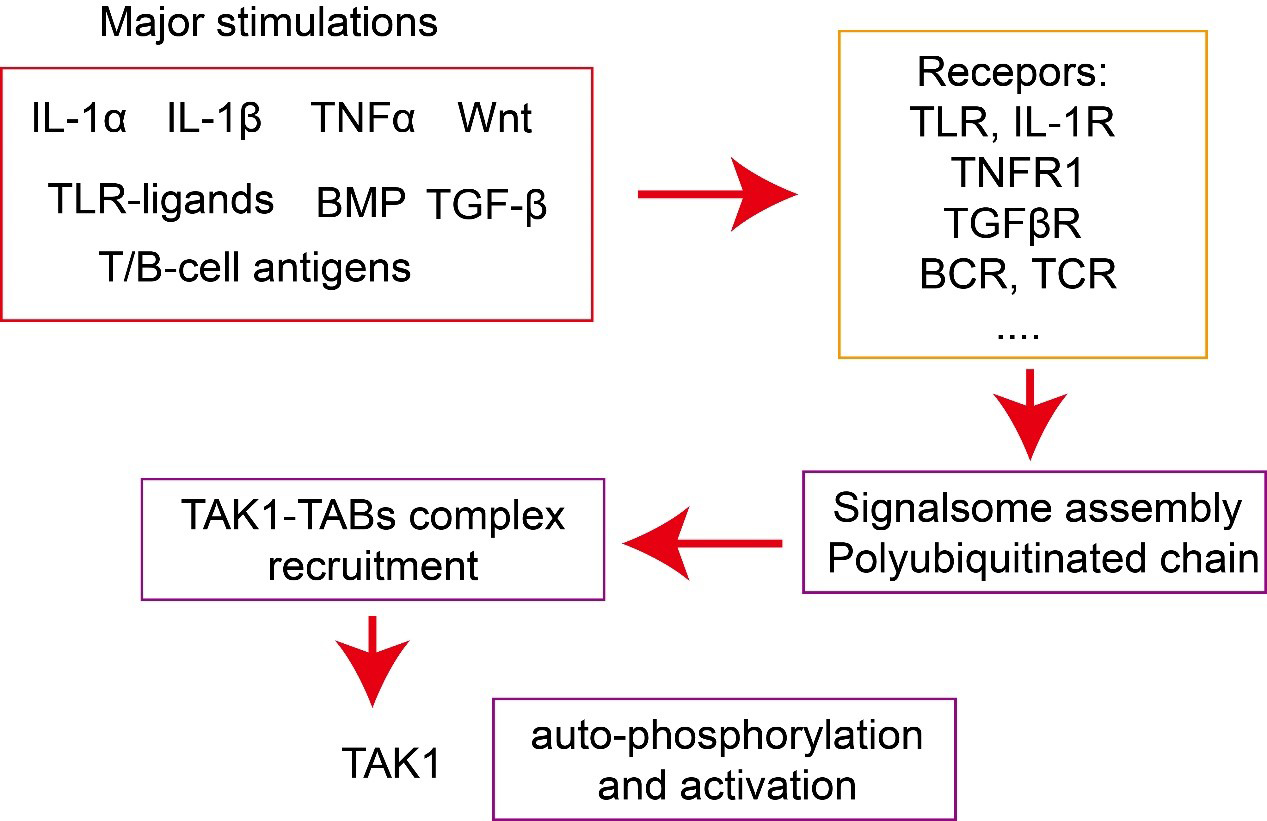 Fig. 2.
Fig. 2.Activation of TAK1–TAB complex. Major stimulations, including
cytokines, such as interleukin (IL)-1
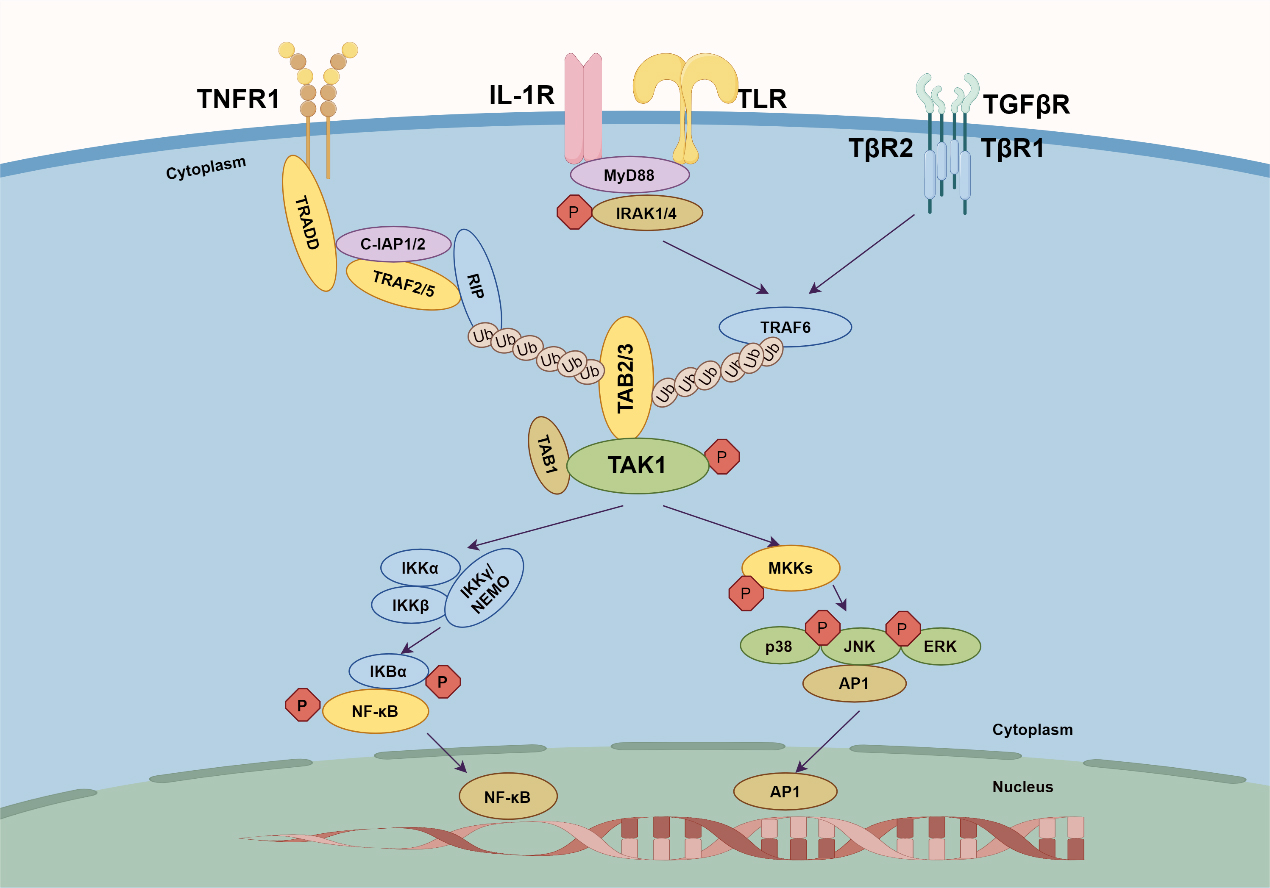 Fig. 3.
Fig. 3.TAK–TAB complex-mediated signaling. Four key receptors are
shown: TNFR1, IL-1R, TLR, and TGF
TNF serves as a central cytokine in inflammatory responses and a common ligand
in the NF-
Recently, GWAS studies have underscored the pivotal role of TNF–TAK1 complex signaling in the pathogenesis of diverse conditions, illuminating the critical function of TAK1 as a therapeutic target. For example, GWAS findings in Asian populations with rheumatoid arthritis (RA) underscore the involvement of MAP3K7 (TAK1) and its activator, TAB1, suggesting a nuanced genetic predisposition [23]. Furthermore, single-nucleotide polymorphisms (SNPs) within the TNF–TAK1 axis have been linked to a heightened risk of inflammatory diseases, including Crohn’s disease and ulcerative colitis, implicating genes such as MAP3K7 and TNFSF18, among others [24, 26, 27]. Despite the advancement of biological therapies targeting TNF, the emergence of resistance highlights the necessity of alternative strategies [39, 40]. The advent of selective TAK1 inhibitors represents a promising avenue, with preclinical studies validating the efficacy of blocking TAK1 in RA [41]. This development not only broadens our therapeutic arsenal but also emphasizes that it is imperative for further research to elucidate the intricate role of TAK1 within inflammatory pathways.
IL-1, also known as lymphocyte-activating factor, is mainly produced by
monocytes/macrophages and exists in two forms: IL-1
Members of the TLR family recognize antigen components of different types of
microorganisms and are well-studied pattern recognition receptors (PRRs). Both
IL-1R and TLR receptors share Toll/interleukin-1 receptor domains in their
intracellular signaling-related structures and, when activated, can bind MyD88
through these Toll/interleukin-1 receptor domains [42, 43, 49]. MyD88 [44],
containing a death domain, then recruits IRAK1/4, TRAF6 and initiates downstream
NF-
The TGF-
Previous research has established TAK1 as a key upstream signaling molecule in
the non-Smad signaling pathway induced by TGF-
Given the crucial role of the TAK1–TAB complex in various signaling pathways responding to diverse immune stimuli, the regulatory cascade mediated by TAK1–TAB has been extensively explored [12]. The TAK1–TAB complex undergoes dynamic post-translational modifications, including phosphorylation, ubiquitination, methylation, acetylation, O-GlcNAcylation, and SUMOylating [16, 58, 59, 60]. These modifications are not mutually exclusive and can interact with each other, further adding to the complexity of regulating the TAK1–TAB complex.
Given the breadth of this topic, the subsequent discussion will focus on
elucidating the role and mechanisms specifically associated with the
ubiquitination of the TAK1–TAB complex. This aspect is particularly critical as
it affects the degradation and recycling of these proteins and profoundly impacts
their function in signaling pathways, such as those leading to NF-
In the context of disease, the dysregulation of TAK1/TAB ubiquitination has been
implicated in the pathogenesis of autoimmune conditions, tumor growth, and
neurodegenerative diseases [8, 21, 61]. The excessive or prolonged activation of
NF-
Ubiquitination, a pivotal post-translational modification, intricately regulates cellular processes critical for maintaining homeostasis [8, 64], including the cell cycle, proliferation, and apoptosis [65]. The ubiquitin–proteasome system (UPS) is crucial for protein degradation, similar to the regulatory roles of kinases [66, 67]. Disruptions in ubiquitination pathways can lead to an imbalance in these processes, significantly contributing to many disorders, including cancer, neurodegenerative diseases, and issues related to adaptive and innate immunity [68, 69]. Recent studies have underscored the dual role of ubiquitination in either promoting or inhibiting tumor progression, depending on the context and the specific proteins being targeted, while the UPS was also demonstrated as a potent target for anticancer therapy [67, 68, 70]. Deubiquitinating enzymes (DUBs) have emerged as critical regulators within this context, capable of removing ubiquitin modifications and, thus, reversing the effects on target proteins. Many DUBs have demonstrated tumorigenic roles in multiple cancers, and many inhibitors have been investigated in clinical trials for diverse cancer treatments [70]. Given their central role in modulating the stability and activity of key proteins involved in tumorigenesis, DUBs represent promising targets for therapeutic intervention, offering the potential for developing novel anticancer strategies [70]. However, the FDA has currently only approved a few drugs that target the ubiquitin system [66].
The advancements in ubiquitin biology have led to innovative strategies for modulating the ubiquitination system, with significant implications for therapeutic development [67]. For the past decade, proteasome inhibitors have been utilized in treating multiple myeloma (MM), yet resistance to these inhibitors has emerged, impacting their effectiveness. Bortezomib (Velcade) and carfilzomib (Kyprolis) are FDA-approved proteasome inhibitors effective against multiple myeloma [66]. Techniques such as proteolysis-targeting chimera (PROTAC) molecules [71] and hydrophobicity tags (HyT) [72] have been developed to specifically target and manipulate the degradation of proteins, offering precise approaches to treat diseases caused by dysregulation of the ubiquitination process [68]. Furthermore, the advent of small molecule inhibitors targeting ubiquitination enzymes presents a promising direction in treating conditions such as cancer, highlighting the potential of these molecular interventions in precision medicine [66].
Ubiquitination and phosphorylation are among the most crucial and widely studied post-translational modifications. It is a dynamic and versatile mechanism for controlling various aspects of cellular function [8, 9]. Ubiquitin, a small yet pivotal protein in cellular regulation, consists of 76 amino acids and is notable for its seven lysine residues—K6, K11, K27, K29, K33, K48, and K63—plus an N-terminal methionine (M1) that can all serve as attachment points for forming polyubiquitin chains [64, 73]. The functions and implications of ubiquitination through K6, K27, K29, and K33 are less well-characterized and represent an area ripe for further research [74, 75]. This ubiquitination system is a key regulator of intracellular proteins, influencing as much as 80% of them [70]. The consequences of ubiquitination can vary greatly (Fig. 4).
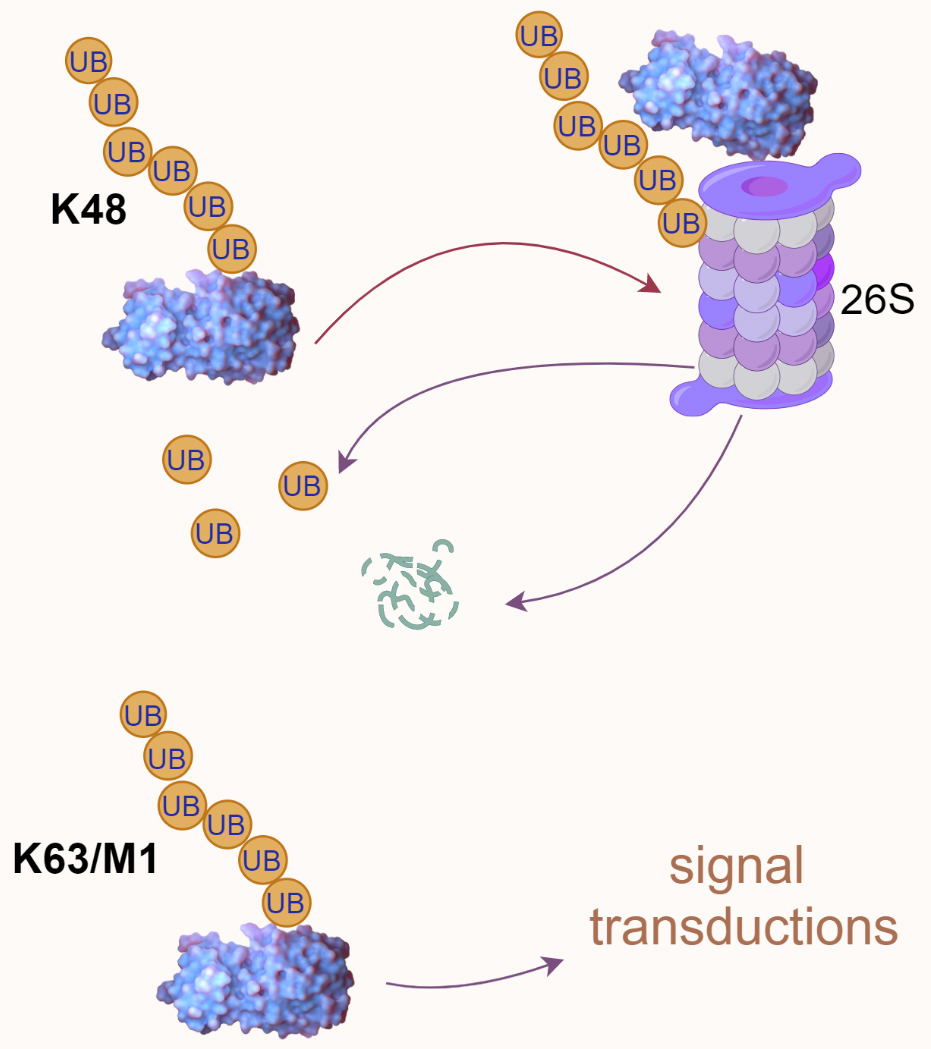 Fig. 4.
Fig. 4.Ubiquitin-mediated proteasomal degradation and signal transduction. K48-linked polyubiquitination is typically associated with proteasomal degradation, as illustrated by a polyubiquitinated protein directed to the 26S proteasome. In contrast, K63/M1-linked polyubiquitination is often involved in signal transduction processes. UB, ubiquitin.
Proteasomal degradation: Ubiquitin chains, particularly those linked
through the K48 residue, often signal the protein to be directed to the 26S
proteasome for degradation. This is a key process for removing damaged,
misfolded, or unneeded proteins, thereby regulating protein levels and quality
control within the cell [73]. Altering substrate activity:
Ubiquitination can also change the functional state of proteins, either
activating or inhibiting their enzymatic activity, altering their cellular
location, or affecting their ability to interact with other molecules [64].
Mediating protein–protein interactions: Ubiquitin chains can serve as a
platform for assembling protein complexes, which is critical in various signaling
pathways. For example, K63-linked ubiquitin chains are often involved in
processes such as DNA repair, NF-
Ubiquitination encompasses non-proteolytic roles such as receptor internalization, multiprotein complex assembly, intracellular trafficking, and key signaling pathways, including those governing inflammation, autophagy, DNA repair, and enzymatic regulation [76, 77, 78]. Deregulation can trigger oncogenic pathways, disrupt cellular metabolism, and lead to inadequate protein complex formation critical for inflammation response or DNA repair, resulting in the accumulation of misfolded proteins, causing diseases such as neurodegeneration or misdirecting proteins away from their functional locations within the cell [8, 79].
Ubiquitination is a multistep process typically involving the coordinated action of three ubiquitination enzymes: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin-protein ligase (E3) [64]. Initially, the E1 forms a high-energy thioester bond between the carboxyl group of the C-terminal lysine residue of ubiquitin and the thiol group of its own cysteine residue, utilizing ATP-provided energy. Subsequently, the activated ubiquitin is transferred onto the cysteine residue of the E2. Finally, a member of the highly conserved ubiquitin-protein ligase family, E3, recognizes specific target proteins to be ubiquitinated and catalyzes the transfer of ubiquitin molecules from E2 to the lysine residue of the target protein (Fig. 5) [64]. Really interesting new gene (RING) finger [RNF] proteins containing RING domains act as E3 ubiquitin ligases that mediate the covalent linkage of ubiquitin to target proteins [9]. Depending on the ubiquitin chain’s topology, ubiquitination can lead to proteasomal degradation or mediate protein–protein interactions, influencing various cellular processes [64]. Understanding these processes provides insights into the molecular mechanisms of diseases involving the TAK1–TAB complex and offers new opportunities for drug development.
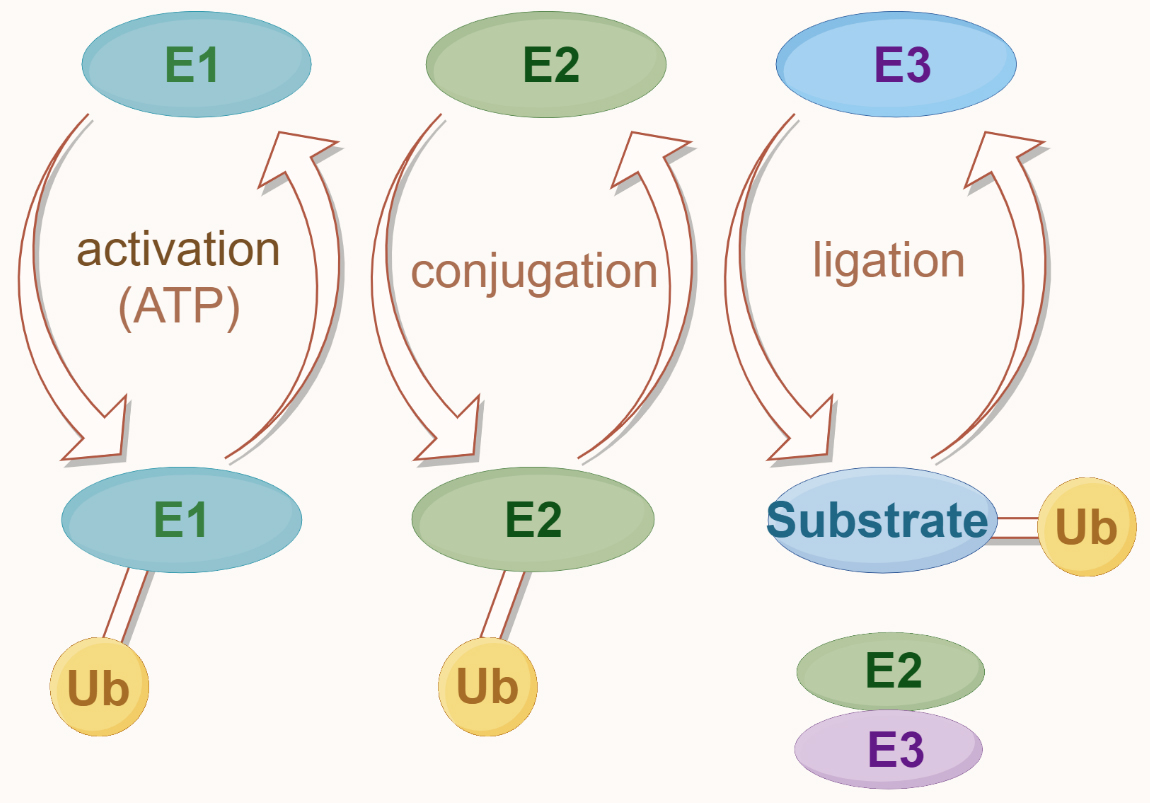 Fig. 5.
Fig. 5.The ubiquitin modification system. Ubiquitination begins with the activation of ubiquitin (Ub) by the E1 enzyme, which is ATP-dependent. Activated ubiquitin is then transferred to the E2 conjugating enzyme. In the final step, the E3 ligase enzyme facilitates the attachment of ubiquitin to the substrate protein, completing the ubiquitination process.
TAK1 undergoes sophisticated regulation via different ubiquitin chain types,
including K63-linked and K48-linked ubiquitination, each dictating unique
biological outcomes [16]. E3 ubiquitin ligases, notably TRAF6 and TRAF2 [80, 81, 82, 83, 84],
are pivotal in this regulation, intricately influencing TAK1’s cellular role.
K63-linked polyubiquitination, identified at lysine residues K34, K158, K209, and
K562, is crucial for TAK1 activation, with TRAF6 and TRAF2 mediation highlighting
a complex regulatory network. A study by Fan et al. [81, 85] revealed
that the activation of TAK1 via NF-
Recent investigations have expanded our understanding of TAK1 activation,
revealing that its function may not solely depend on ubiquitination at known
sites, suggesting a labyrinth of regulatory pathways yet to be explored. The tripartite motif-containing (TRIM)
protein family [9], notably TRIM8 [86, 87] and TRIM27 [88],
emerges as significant modulators within the TAK1/NF-
These investigations suggest that TAK1 activation is intricately modulated beyond known ubiquitination sites, hinting at a broader spectrum of regulatory pathways awaiting discovery. The involvement of the TRIM protein family, among other E3 ligases, in modulating TAK1 through diverse ubiquitination patterns underscores the multifaceted nature of its regulation. This complexity is crucial for maintaining cellular homeostasis and highlights the potential for targeted therapeutic interventions in diseases associated with TAK1 pathway dysregulation (Table 1, Ref. [57, 61, 80, 81, 82, 83, 84, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104]).
| Protein | Catalyzed by | Site | PTM | Reference |
| TAK1 | TRAF6 | K34, K158, K562 | K63 Ub | [80, 81, 82, 83, 84] |
| TRAF2 | K158 | K63 Ub | [82] | |
| TRIM8 | K158 | K63 Ub | [86, 87] | |
| Sef-S | K209 | K63 Ub | [89] | |
| CHIP | K63 Ub | [90] | ||
| ITCH | K72 | K48 Ub | [88, 92] | |
| TRIM31 | K72 | K48 Ub | [57] | |
| TRIM16 | K282, 547 | K48 Ub | [91] | |
| FBXW2 | K48 Ub | [93] | ||
| TRIM56 | M1 Ub | [94] | ||
| TAB1 | PHD | K294, K319, K335, K350 | K63 Ub | [61] |
| RNF207 | K63 Ub | [95] | ||
| ITCH | S452, S453, S456, S457 | K48 Ub | [96] | |
| TRIM26 | K294, K319 | K11 Ub | [97] | |
| K335 | ||||
| RNF114 | Ub | [98] | ||
| TAB2 | TRIM29 | K48 Ub | [99] | |
| RBCK1 | C673 | K48 Ub | [100] | |
| RNF99 | K611 | K48 Ub, | [101, 102] | |
| K63 Ub | ||||
| TRIM23 | Ub | [103] | ||
| TAB3 | AMFR | K649 | K27 Ub | [104] |
| RBCK1 | C692 | K48 Ub | [100] |
PTM, post-translational modification; K, lysine; C, cysteine; Ub, ubquitination.
TAB1, a critical regulator of TAK1 activity through direct interaction, is subject to a complex regulatory network via polyubiquitination and other post-translational modifications, including phosphorylation and O-GlcNAcylation [105, 106]. This multilayered modification landscape underscores the pivotal role of TAB1 in cellular signaling dynamics. Notably, both K63 and K48-linked ubiquitination have been observed on TAB1 [61, 95, 96], highlighting its versatile regulatory capabilities.
K63-linked ubiquitination of TAB1, particularly at lysine residues Lys294, Lys319, Lys335, and Lys350, is mediated by various E3 ligases, including the E3 ubiquitin ligase activity of the mitogen-activated protein kinase kinase 1 (MEKK1) the plant homeodomain (PHD) [61]. This modification is crucial for enhancing TAK1 aggregation and its kinase activity, significantly influencing embryonic stem cell differentiation and tumorigenesis. Intriguingly, recent discoveries, such as the work by Yuan et al. [95], have illuminated the role of RNF207 in activating the TAK1–JNK1/2 signaling pathway through K63-linked ubiquitination of TAB1, linking it to the exacerbation of stress-induced pathological cardiac hypertrophy.
TAB1 can also undergo K48-linked ubiquitination, although the specific
modification sites remain unclear. Immune regulation is essential for maintaining
skin integrity. Research by Theivanthiran et al. [96] suggests that
ITCH can catalyze K48-linked polyubiquitination on TAB1, inhibiting the
activation of p38
In addition to K63 and K48-linked ubiquitination, other types of ubiquitination
also play crucial roles in TAB1’s functions. In a study by Zhao et al.
[97], TRIM26 was found to catalyze K11-linked polyubiquitination on
TAB1 at positions Lys294, Lys319, and Lys335, enhancing the activation of TAK1
and subsequent NF-
This enriched understanding of TAB1’s ubiquitination broadens our perspective on its regulatory functions and underscores the potential for targeting these modifications in therapeutic strategies, especially in contexts of cellular stress responses and disease pathogenesis.
TAB2 and TAB3 recognize and bind to ubiquitinated substrates and undergo
self-ubiquitination, thereby finely tuning the intensity and duration of signal
transduction [12, 107]. Human TAB2 and TAB3 feature two conserved
ubiquitin-binding domains: the N-terminal CUE and C-terminal NZF domains. The CUE
domain, a universal ubiquitin-binding structure, includes the
phenylalanine–proline (Phe–Pro or FP) motif
crucial for ubiquitin-binding [33, 108]. Although the precise role of the CUE
domain in TAB2 and TAB3 remains unclear, studies suggest that the absence of this
domain partially reduces the ability of TAB2 and TAB3 to activate NF-
The ubiquitin-modified TAB2 and TAB3 primarily serve two functions. First, the NZF domain within TAB2 and TAB3 facilitates the binding to K63-linked polyubiquitin chains on partner proteins such as RIP and TRAF6 [33]. This interaction is crucial for activating TAK1, leading to the regulation of downstream signaling cascades that govern immune and inflammatory responses, cellular autophagy, and apoptotic processes [62]. Conversely, ubiquitination can signal the degradation of TAB2 and TAB3, thus attenuating inflammatory responses. Theoretically, both proteins can be targeted for degradation within the cell through their NZF domains, which can also bind to K48-linked polyubiquitin chains—the classic marker for proteasomal degradation [33, 109]. However, current research suggests that TAB2 and TAB3 have a higher binding affinity for K63 chains over K48 chains [62, 107]. Notably, TAB3 exhibits a significantly lower affinity for K48 chains than TAB2, indicating that TAB2 may be more prone to degradation via the K48 ubiquitination–proteasome pathway. This differential affinity for ubiquitin chains suggests distinct regulatory mechanisms for TAB2 and TAB3, influencing their stability and function in cellular signaling pathways [62, 107].
Their modification significantly influences the functionality of TAB2 and TAB3
through various E3 ubiquitin ligases, such as TRAF6 and members of the TRIM
protein family. Upon stimulation with cytokines such as TNF
Other E3 ligases, such as RNF4, have also been implicated in the lysosomal degradation of TAB2 or TAB3. Moreover, selective autophagy, particularly mediated by the autophagy receptor NBR1, is another pathway through which TAB3 may be degraded [115]. Qiu’s [111] research has presented that TRIM22 can interact with TAB2 independently of its RING domain and lead to the degradation of TAB2 via the proteasome pathway. Nevertheless, the necessity of TAB2 ubiquitination for TRIM-mediated degradation is yet to be conclusively determined, and further experimental research is needed to validate the specific modifications of TAB2.
K48-linked protein ubiquitination modification directs degradation through the
proteasome pathway [64]. It has been demonstrated that TRIM29 catalyzes
substrate ubiquitination through its B-box domain rather than the typical RING
domain. Dou’s [99] study found that TRIM29 in NK cells can bind to TAB2,
mediating ubiquitination and proteasome degradation of Lys48-linked TAB2, thereby
inhibiting the production of IFH-
A recent study has indicated a potential link between TRIM23 and TAB2 ubiquitination in macrophages and the alleviation of kidney symptoms in diabetic mice. However, further verification is required to determine whether TRIM23 directly mediates the ubiquitination of TAB2. Nonetheless, the direct involvement of TRIM23 in the ubiquitination process of TAB2 remains to be conclusively established [103].
Sun’s [104] study on the ubiquitination of TAB3 reveals that the autocrine motility factor receptor (AMFR), an E3 ubiquitin ligase anchored to the endoplasmic reticulum (ER) in an ER-dependent and loop-dependent manner, directly interacts with TAB3, inducing K27-linked polyubiquitination of TAB3 at Lys649 and promoting TAK1 activation.
Emerging research points to a diverse landscape of ubiquitination-induced regulatory mechanisms for TAB2 and TAB3, implicating various E3 ligases in their modification. This diversity underscores the complexity of cellular signaling regulation and the potential of targeting these pathways for therapeutic intervention. Further investigations into the specific roles and interaction sites of E3 ligases will undoubtedly deepen our understanding of TAB2 and TAB3 regulation, paving the way for novel insights into cellular dynamics and disease pathogenesis.
The activation of NF-
Several DUBs, each with distinct roles, are instrumental in regulating the TAK1–TAB complex. By selectively removing ubiquitin chains from these proteins, these enzymes can alter the activity, stability, and interaction of TAK1 and TABs, thereby fine-tuning the signaling pathways they are involved in [118, 121, 122, 123]. This targeted deubiquitylation is crucial for precisely controlling signaling events that govern cellular processes such as inflammation, cell survival, and proliferation, with implications for both normal physiology and disease states, including cancer [63].
Enzymes such as USP4, USP18, USP19, and CYLD [118, 121, 122, 123], along with members of the TRIM family, such as TRIM44 [124] and TRIM15 [125], are known to modulate the functions of the TAK1 and TAB proteins through their specific deubiquitinating activities (Table 2, Ref. [92, 116, 117, 118, 121, 122, 123, 124, 125, 126, 127, 128]).
| Protein | Catalyzed by | Site | PTM | Reference |
| TAK1 | USP4 | K158 | K63 de-Ub | [116, 121, 126] |
| USP8 | K63 de-Ub | [127] | ||
| CYLD | K209 | K63 de-Ub | [92] | |
| USP18 | K63 de-Ub | [117, 118] | ||
| USP19 | K63 de-Ub | [123] | ||
| K27 de-Ub | ||||
| TRIM15 | K63 de-Ub | [125] | ||
| TRIM44 | K48 de-Ub | [124] | ||
| TAB2 | USP15 | K48 de-Ub | [122] | |
| USP14 | de-Ub | [128] |
PTM, post-translational modification; K, lysine; C, cysteine; Ub, ubquitination; de-Ub, Deubiquitination.
USP4: Inhibits the activation of NF-
The exploration of DUBs such as USP4, USP18, USP19, and CYLD, in conjunction with TRIM family members such as TRIM44 and TRIM15, underscores their pivotal roles in modulating the function and regulation of the TAK1–TAB signaling complex [60, 116, 117, 118]. These enzymes orchestrate a delicate balance within cellular signaling pathways, particularly those implicated in inflammation and immune responses. By engaging in specific deubiquitinating actions, these DUBs and TRIM proteins contribute to a sophisticated regulatory network that maintains cellular homeostasis and modulates the body’s response to various stresses and pathological states [8].
The stability of TAB2 and TAB3 is vital for cellular homeostasis, regulated by a balance between their degradation and stabilization through lysosomal and proteasomal pathways [5, 6, 7, 16]. This equilibrium is influenced by ubiquitination, marking the proteins for degradation or protection, crucial for their roles in inflammation, immunity, and apoptosis [129]. Understanding how this balance is maintained can reveal insights into cellular signaling and offer new targets for treating diseases related to pathway dysregulation. Further research into these mechanisms is essential for advancing our knowledge of cellular processes and developing therapeutic interventions.
In a notable advancement, Zhou et al. [122] identified that
USP15, a deubiquitinating enzyme, is critical in regulating TAB2 by cleaving
K48-linked ubiquitin chains. This action by USP15 curtails the degradation of
TAB2, which would typically occur via lysosomal pathways, thereby enhancing the
stability of TAB2 and contributing to the subsequent NF-
Furthermore, ubiquitin-specific protease 14 (USP14) [128], which is
known to be a significant player in proteasomal degradation, has been implicated
in the development and progression of various cancers. Research indicates that
USP14 can interact with TRAF6 without altering its ubiquitination status. Yet, it
can promote the deubiquitination of TAB2, a known substrate of TRAF6. This action
attenuates NF-
Deubiquitinating enzymes play a crucial role in the NF-
Within the MAP3K family, TAK1 is crucial in orchestrating signaling pathways for
inflammatory responses, interacting closely with TAB proteins to significantly
influence the NF-
The dysregulation of the ubiquitination of the TAK–TAB complex is linked to the onset of numerous diseases, from chronic inflammation to cancer, highlighting the importance of understanding these mechanisms for developing targeted therapies. Enhancing our knowledge in this area could lead to groundbreaking drug discoveries. Research challenges include precisely controlling the activity of the TAK1–TAB complex and avoiding potential side effects of interventions. Thus, a deeper investigation into the functional mechanisms of TAK1 is vital for crafting more effective treatments. Furthermore, this regulation through ubiquitination underscores the broader significance of post-translational modifications in cellular signaling balance, promising advancements in therapeutic innovations through continued exploration.
The progress in bioinformatics, mass spectrometry, and high-throughput screening technologies has made developing specific E3 inhibitors for the TAK1–TAB complex more achievable. Techniques such as phage display have been utilized to create ubiquitin variants that can specifically inhibit E3 ligases and DUBs [130]. Moreover, computer-aided drug design has led to innovative therapeutic strategies that can disrupt E3 ligase-substrate interactions [66]. The advent of degrader technologies such as PROTACs, HyTs, and selective estrogen receptor degraders (SERDs) opens new avenues for drug development in targeting the TAK1–TAB complex, potentially leading to significant clinical advancements.
MZ, YZ and CZ contributed to the conception and design of the work. JZ, LC, LJL, WQQ, WY, CYK and RQR collected the literature and information to write the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript. And all authors read and approved the final version of the manuscript.
Not applicable.
Not applicable.
This work was supported by grants of the National Natural Science Foundation of China (No. 82270487).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.